Publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2026
- CELL REP
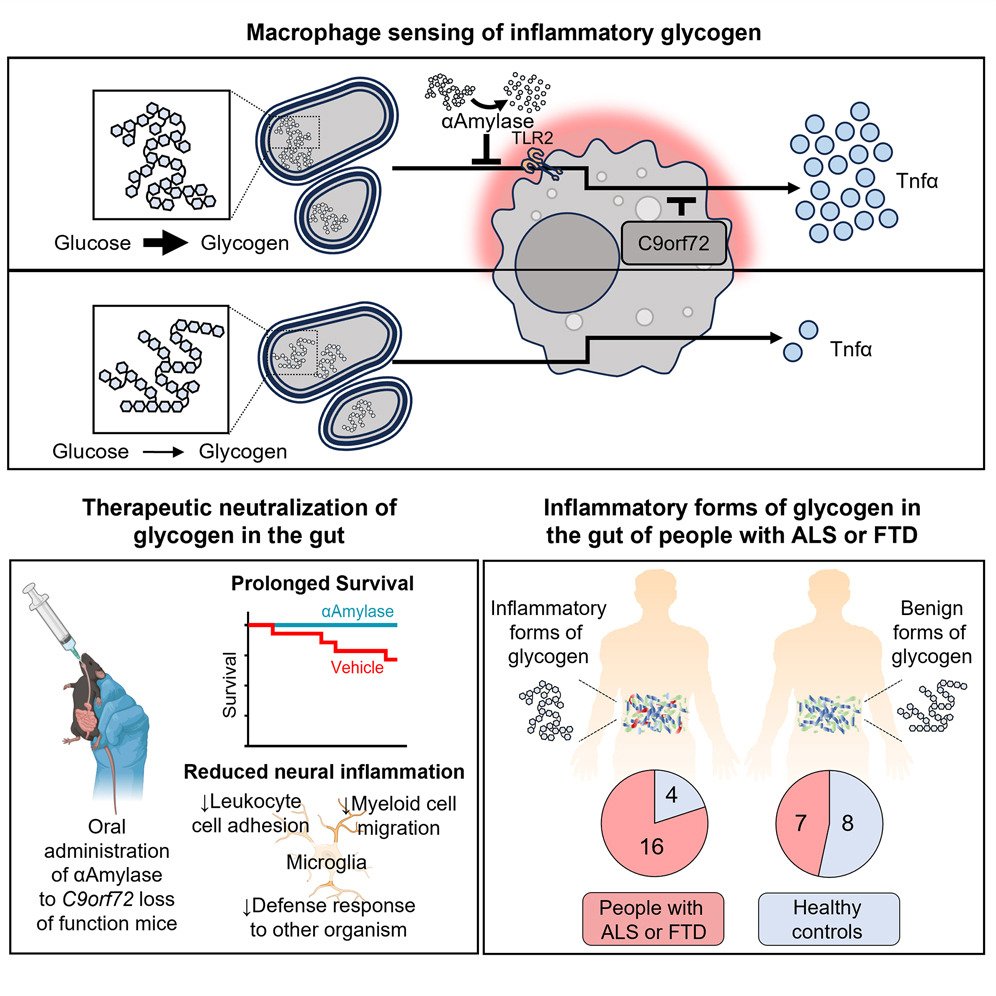 C9orf72 in myeloid cells prevents an inflammatory response to microbial glycogenBlake McCourt, Katelyn Lemr, Shinjon Chakrabarti, Elizabeth Woidke, Sara Ramaiah, Vaidhvi Singh, Naseer Sangwan, J. Mark Brown, Fabio Conminelli, Alex Rodriguez-Palacios, and Aaron BurberryCell Reports, Feb 2026
C9orf72 in myeloid cells prevents an inflammatory response to microbial glycogenBlake McCourt, Katelyn Lemr, Shinjon Chakrabarti, Elizabeth Woidke, Sara Ramaiah, Vaidhvi Singh, Naseer Sangwan, J. Mark Brown, Fabio Conminelli, Alex Rodriguez-Palacios, and Aaron BurberryCell Reports, Feb 2026Gut dysbiosis and neural inflammation occur in patients with amyotrophic lateral sclerosis (ALS), including those with a causal mutation in chromosome 9 open reading frame 72 (C9ORF72). How gut commensals interact with common ALS genotypes to impart risk of neural degeneration remains unclear. Here, we identify 10 phylogenetically diverse bacterial strains that promote cytokine release in a C9orf72-dependent manner. Metatranscriptomics implicated the glycogen biosynthesis pathway as a driver of inflammation. Colonization of germ-free C9orf72-deficient mice with Parabacteroides merdae that produced inflammatory glycogen enhanced monocytosis, blood-brain barrier breakdown, and T cell infiltration into the central nervous system. Enzymatic digestion of glycogen in the gut promoted survival of C9orf72-deficient mice and dampened microglial reactivity in the brain. A survey of human fecal samples demonstrated that inflammatory forms of glycogen were present in gut contents from 15/22 patients with ALS, 1/1 patient with C9ORF72 frontotemporal dementia (FTD), and 4/12 healthy controls. Together, the results of this work identify bacterial glycogen as a modifiable mediator of immune homeostasis in the gut and brain.
@article{cellreports2026glycogen, author = {McCourt, Blake and Lemr, Katelyn and Chakrabarti, Shinjon and Woidke, Elizabeth and Ramaiah, Sara and Singh, Vaidhvi and Sangwan, Naseer and Brown, J. Mark and Conminelli, Fabio and Rodriguez-Palacios, Alex and Burberry, Aaron}, title = {C9orf72 in myeloid cells prevents an inflammatory response to microbial glycogen}, journal = {Cell Reports}, volume = {45}, number = {}, pages = {}, year = {2026}, month = feb, doi = {10.1016/j.celrep.2025.116906}, url = {https://www.cell.com/cell-reports/fulltext/S2211-1247(25)01678-X}, eprint = {}, }

2024
- Myeloid and lymphoid expression of C9orf72 regulates IL-17A signaling in miceFrancesco Limone, Alexander Couto, Jin-Yuan Wang, Yingying Zhang, Blake McCourt, Cerianne Huang, Adina Minkin, Marghi Jani, Sarah McNeer, James Keaney, Gaëlle Gillet, Rodrigo Lopez Gonzalez, Wendy A. Goodman, Irena Kadiu, Kevin Eggan, and Aaron BurberryScience Translational Medicine, Feb 2024
A mutation in C9ORF72 is the most common cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Patients with ALS or FTD often develop autoimmunity and inflammation that precedes or coincides with the onset of neurological symptoms, but the underlying mechanisms are poorly understood. Here, we knocked out murine C9orf72 in seven hematopoietic progenitor compartments by conditional mutagenesis and found that myeloid lineage C9orf72 prevents splenomegaly, loss of tolerance, and premature mortality. Furthermore, we demonstrated that C9orf72 plays a role in lymphoid cells to prevent interleukin-17A (IL-17A) production and neutrophilia. Mass cytometry identified early and sustained elevation of the costimulatory molecule CD80 expressed on C9orf72-deficient mouse macrophages, monocytes, and microglia. Enrichment of CD80 was similarly observed in human spinal cord microglia from patients with C9ORF72-mediated ALS compared with non-ALS controls. Single-cell RNA sequencing of murine spinal cord, brain cortex, and spleen demonstrated coordinated induction of gene modules related to antigen processing and presentation and antiviral immunity in C9orf72-deficient endothelial cells, microglia, and macrophages. Mechanistically, C9ORF72 repressed the trafficking of CD80 to the cell surface in response to Toll-like receptor agonists, interferon-γ, and IL-17A. Deletion of Il17a in C9orf72-deficient mice prevented CD80 enrichment in the spinal cord, reduced neutrophilia, and reduced gut T helper type 17 cells. Last, systemic delivery of an IL-17A neutralizing antibody augmented motor performance and suppressed neuroinflammation in C9orf72-deficient mice. Altogether, we show that C9orf72 orchestrates myeloid costimulatory potency and provide support for IL-17A as a therapeutic target for neuroinflammation associated with ALS or FTD. The ALS-associated C9orf72 gene product opposes IL-17A–dependent inflammation in myeloid and lymphoid cells. Patients with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) often exhibit autoimmune inflammation before neurological symptoms. Although mutated C9ORF72 has been linked to ALS and FTD, its role in neuroinflammation is unclear. Limone et al. found that hematopoietic loss of C9orf72 expression drove excess IL-17A inflammation. Myeloid-specific loss of C9orf72 was sufficient to cause severe autoimmunity. C9orf72-deficient mice had more myeloid cells with higher surface expression of costimulatory molecule CD80, which was potentiated by IL-17A. Patients with C9ORF72-related ALS similarly showed CD80 enrichment in spinal cord microglia. IL-17A neutralizing antibody reduced neuroinflammation in C9orf72-deficient mice, supporting further investigation of IL-17A–based therapies for ALS- and FTD-associated neuroinflammation. —Molly Ogle
@article{scitm2024il17a, author = {Limone, Francesco and Couto, Alexander and Wang, Jin-Yuan and Zhang, Yingying and McCourt, Blake and Huang, Cerianne and Minkin, Adina and Jani, Marghi and McNeer, Sarah and Keaney, James and Gillet, Gaëlle and Gonzalez, Rodrigo Lopez and Goodman, Wendy A. and Kadiu, Irena and Eggan, Kevin and Burberry, Aaron}, title = {Myeloid and lymphoid expression of <i>C9orf72</i> regulates IL-17A signaling in mice}, journal = {Science Translational Medicine}, volume = {16}, number = {732}, pages = {eadg7895}, year = {2024}, doi = {10.1126/scitranslmed.adg7895}, url = {https://www.science.org/doi/abs/10.1126/scitranslmed.adg7895}, eprint = {https://www.science.org/doi/pdf/10.1126/scitranslmed.adg7895}, }

2020
- C9orf72 suppresses systemic and neural inflammation induced by gut bacteriaAaron Burberry, Michael F. Wells, Francesco Limone, Alexander Couto, Kevin S. Smith, James Keaney, Gaëlle Gillet, Nick Gastel, Jin-Yuan Wang, Olli Pietilainen, and al.Nature, May 2020
A hexanucleotide-repeat expansion in C9ORF72 is the most common genetic variant that contributes to amyotrophic lateral sclerosis and frontotemporal dementia1,2. The C9ORF72 mutation acts through gain- and loss-of-function mechanisms to induce pathways that are implicated in neural degeneration3,4,5,6,7,8,9. The expansion is transcribed into a long repetitive RNA, which negatively sequesters RNA-binding proteins5 before its non-canonical translation into neural-toxic dipeptide proteins3,4. The failure of RNA polymerase to read through the mutation also reduces the abundance of the endogenous C9ORF72 gene product, which functions in endolysosomal pathways and suppresses systemic and neural inflammation6,7,8,9. Notably, the effects of the repeat expansion act with incomplete penetrance in families with a high prevalence of amyotrophic lateral sclerosis or frontotemporal dementia, indicating that either genetic or environmental factors modify the risk of disease for each individual. Identifying disease modifiers is of considerable translational interest, as it could suggest strategies to diminish the risk of developing amyotrophic lateral sclerosis or frontotemporal dementia, or to slow progression. Here we report that an environment with reduced abundance of immune-stimulating bacteria10,11 protects C9orf72-mutant mice from premature mortality and significantly ameliorates their underlying systemic inflammation and autoimmunity. Consistent with C9orf72 functioning to prevent microbiota from inducing a pathological inflammatory response, we found that reducing the microbial burden in mutant mice with broad spectrum antibiotics—as well as transplanting gut microflora from a protective environment—attenuated inflammatory phenotypes, even after their onset. Our studies provide further evidence that the microbial composition of our gut has an important role in brain health and can interact in surprising ways with well-known genetic risk factors for disorders of the nervous system.
@article{nature2020bacteria, author = {Burberry, Aaron and Wells, Michael F. and Limone, Francesco and Couto, Alexander and Smith, Kevin S. and Keaney, James and Gillet, Gaëlle and van Gastel, Nick and Wang, Jin-Yuan and Pietilainen, Olli and et al.}, title = {C9orf72 suppresses systemic and neural inflammation induced by gut bacteria}, journal = {Nature}, volume = {582}, number = {7810}, pages = {89-94}, year = {2020}, month = may, doi = {10.1038/s41586-020-2288-7}, }

2016
- Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune diseaseAaron Burberry, Naoki Suzuki, Jin-Yuan Wang, Rob Moccia, Daniel A. Mordes, Morag H. Stewart, Satomi Suzuki-Uematsu, Sulagna Ghosh, Ajay Singh, Florian T. Merkle, Kathryn Koszka, Quan-Zhen Li, Leonard Zon, Derrick J. Rossi, Jennifer J. Trowbridge, Luigi D. Notarangelo, and Kevin EgganScience Translational Medicine, May 2016
Loss-of-function mutations in the mouse ortholog of C9ORF72 cause fatal autoimmunity that is transferable by bone marrow–derived cells, demonstrating a hematopoietic intrinsic function for the protein encoded by this gene. Mutations in C9ORF72 are a common contributor to amyotrophic lateral sclerosis and frontotemporal dementia, yet the function of this gene is still poorly defined. In new work, Burberry et al. demonstrate that mutations disrupting the normal function of C9orf72 cause mice to develop features of autoimmunity. They further found that transplantation of normal bone marrow into mutant animals ameliorated this phenotype, whereas transplantation of mutant bone marrow into normal animals was sufficient to cause autoimmunity. The authors conclude that C9orf72 acts through hematopoietic cells to maintain normal immune function and suggest that investigations are warranted into whether disruptions in immunity contribute to disease in patients. C9ORF72 mutations are found in a significant fraction of patients suffering from amyotrophic lateral sclerosis and frontotemporal dementia, yet the function of the C9ORF72 gene product remains poorly understood. We show that mice harboring loss-of-function mutations in the ortholog of C9ORF72 develop splenomegaly, neutrophilia, thrombocytopenia, increased expression of inflammatory cytokines, and severe autoimmunity, ultimately leading to a high mortality rate. Transplantation of mutant mouse bone marrow into wild-type recipients was sufficient to recapitulate the phenotypes observed in the mutant animals, including autoimmunity and premature mortality. Reciprocally, transplantation of wild-type mouse bone marrow into mutant mice improved their phenotype. We conclude that C9ORF72 serves an important function within the hematopoietic system to restrict inflammation and the development of autoimmunity.
@article{scitm2016c9lof, author = {Burberry, Aaron and Suzuki, Naoki and Wang, Jin-Yuan and Moccia, Rob and Mordes, Daniel A. and Stewart, Morag H. and Suzuki-Uematsu, Satomi and Ghosh, Sulagna and Singh, Ajay and Merkle, Florian T. and Koszka, Kathryn and Li, Quan-Zhen and Zon, Leonard and Rossi, Derrick J. and Trowbridge, Jennifer J. and Notarangelo, Luigi D. and Eggan, Kevin}, title = {Loss-of-function mutations in the <i>C9ORF72</i> mouse ortholog cause fatal autoimmune disease}, journal = {Science Translational Medicine}, volume = {8}, number = {347}, pages = {347ra93-347ra93}, year = {2016}, doi = {10.1126/scitranslmed.aaf6038}, url = {https://www.science.org/doi/abs/10.1126/scitranslmed.aaf6038}, eprint = {https://www.science.org/doi/pdf/10.1126/scitranslmed.aaf6038}, }
